
In December of 2022, FDA released its latest guidance on human factors in medical device marketing submissions. The purpose of the new document is to complement and provide some clarifications to the existing guidance document, Applying Human Factors and Usability Engineering to Medical Device. Here are the most notable updates to the FDA guidance.
More focus on the entire HFE Process
The HF/UE report should include more than just the results of the human factors validation study. Instead, it should describe in detail how the human factors engineering process was applied to the device. For example, it should describe the:
- device’s users
- use environments
- use cases
- presence/absence of critical tasks
- analysis of known risks
- summary of other formative analyses
- validation testing for risk mitigation strategies and
- discussion of residual risks.
DeNovo and Humanitarian Exemption
The new guidance document explicitly includes De Novo, and humanitarian exemption (HDE) applications, whereas the 2016 guidance only references 510(k) and PMA submissions. Actually, the guidance has applied to De Novo and HDE applications for some time, but this new document makes it explicit. As always, human factors may also be relevant to other submissions such as Investigational Device Exemptions (IDEs) even though the new guidance does not mention them.
Definition of Additional Terms
FDA left many of its definitions from the 2016 guidance untouched, however, it did define some additional terms such as:
- Harm (leveraging ANSI/AAMI/ISO 14971): Injury or damage to the health of people, or damage to property or the environment.
- Normal use (per IEC 62366): Operation, including routine inspection and adjustments by any user, and stand-by, according to the instructions for use in accordance with generally accepted practice for those medical devices provided without instructions for use.
- Residual risk (per ANSI/AAMI/ISO 14971): Risk remaining after risk control measures have been implemented.
- Serious harm: Includes both serious injury and death.
- Serious injury: An injury or illness that is life-threatening, results in permanent impairment of a body function or permanent damage to body structure, or necessitates medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure. Permanent means irreversible impairment or damage to a body structure or function, excluding trivial impairment or damage.
Definitions help. For example, the definition of harm helps to emphasize human factors engineering’s role in risk management. Additionally, the 2016 guidance discussed residual risk, but never actually defined it. “Serious harm” and “serious injury” definitions will also be helpful.
The definition of a “critical task” includes tasks that would or could cause serious harm if performed incorrectly or not at all, but not all manufacturers had the same definition of “serious harm.” This will help manufacturers be more consistent with FDA’s definition of what harm is “serious” or “not serious.” Specific definitions help ensure that people are actually discussing the same concept.
Three Submission Categories
FDA describes three medical device submission categories determined by:
- Whether the device is new or a modified form an existing product
- What the change is, if applicable
- Whether critical tasks are involved or affected, or not
These are summarized here.

By using the descriptions, manufacturers can determine what human factors information is needed for their submission. FDA describes exactly what should be discussed in the HF submission for each of the three categories:
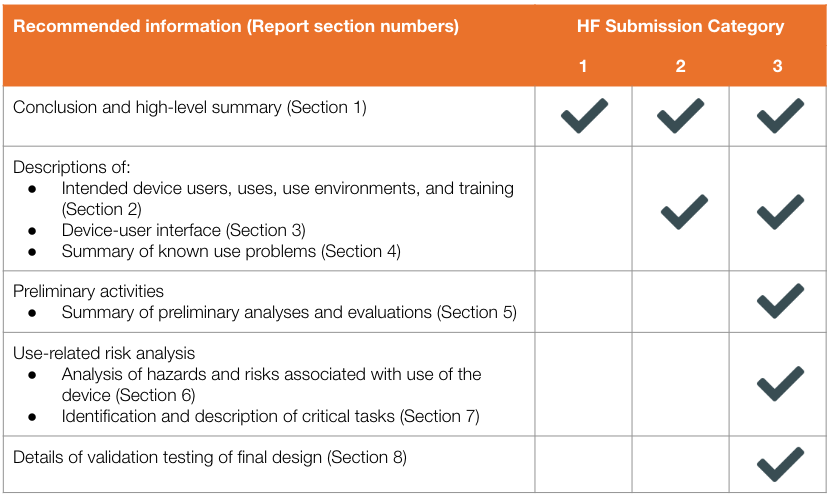
All three submissions require a conclusion and high-level summary of their human factors activities (Section 1 only). Category 2 submissions require additional human factors information such as descriptions of intended device users, uses, use environments, and training (Section 2), device-user interface (Section 3), and summaries of known use problems (Section 4). As the only category with critical tasks involved, only Category 3 requires Sections 5-8.
Recommended content of human factors information in marketing submissions
The newest guidance then walks through what the Agency expects to see in each of the eight sections of the HF/UE report. Below are some notable differences from the 2016 guidance:
Section 1: Conclusion and high-level summary
The contents of Section 1 will depend on which submission category is applicable. For the most part, however, there are no major changes.
Section 2: Descriptions of intended device users, uses, use environments, and training
No major changes
Section 3: Description of device-user interface
If the submission is a modified device, the Agency wants manufacturers to explicitly describe the difference(s) between the existing device and the modified device, including images. For submissions of new devices, there are no major changes.
Section 4: Summary of known use problems
No major changes
Section 5: Summary of preliminary analyses and evaluations
According to the 2016 guidance, the summary of preliminary analyses was documented in Section 6. This order of sections was always rather unintuitive, given that it disrupted the flow between Section 5 (Analysis of hazards and risks) and Section 7 (Identification and description of critical tasks), which are related to each other. The 2022 guidance does not recommend any new/different content in this section; it merely rearranges the order of sections to be more intuitive.
Section 6: Analysis of hazards and risks associated with the use of the device
In the 2016 guidance, FDA recommended referencing a comprehensive risk analysis that contains the use-related hazards and risks. They are now more specifically leaning into the use-related risk analysis, or URRA. This is not necessarily new or different, just a more specific recommendation for how to document the HFE process. The Agency even provides an example table for URRA.
Section 7: Identification and description of critical tasks
The new guidance recommends that the manufacturer describe the levels of severity of a given harm, such as a qualitative five-level severity rating from ISO 14971. This helps clarify how a task’s severity of harm relates to how critical tasks are determined to be “critical.”
Additionally, FDA recommends that manufacturers explicitly (separately) document any new critical tasks, if the submission is for a modified device.
Section 8: Details of HF validation testing of final design
No major changes
Conclusion
The new human factors guidance includes notable changes to the Agency’s current thinking on human factors and usability. For most manufacturers, primarily regulatory personnel, the most important information relates to the submission categories and what information should be included in HF/UE reports per category. However, while seemingly minor details, FDA’s updates to the definitions and the specific guidance on what content should be included in each section are useful.
Contact us to learn more about FDA’s human factors guidance documents and how we can assist your human factors needs.


